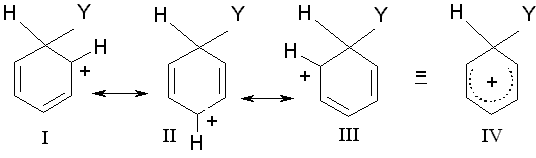
Abbildung 1: Grenzstrukturen und Resonanzhybrid des Carbenium-Ions
Bei der Erst-Substitution am Benzol sind alle 6 C-Atome gleichberechtigt einem elektrophilen Angriff ausgesetzt, deswegen gibt es auch nur ein Monobrombenzol, ein Monochlorbenzol usw. Anders verhält es sich bei der Einführung eines zweiten Substituenten: Jeder Ringsubstituent beeinflusst die Reaktivität des Ringes und die Orientierung, d.h. den Eintrittsort des Zweit-Substituenten in den Ring. Der erste Substituent bestimmt also, wie schnell und wo die zweite Substitution erfolgt.
1. Begriffsbestimmungen
Ein Substituent, der die Reaktivität des Rings im Vergleich zum Benzol erhöht, wird als aktivierend bezeichnet. Umgekehrt erniedrigt ein desaktivierender Substituent die Reaktivität des Ring im Vergleich mit dem Benzol.
Ein Substituent, der eine Zweit-Substitution in ortho- und para-Stellung erleichtert, wird als ortho-para-dirigierend bezeichnet, eine Orientierung in meta-Stellung als meta-dirigierend benannt.
2. Experimentelle Ergebnisse
Aus vergleichenden Untersuchungen, auf die hier nicht weiter eingegangen werden soll, kann geschlossen werden, welche Erst-Substituenten einen aktivierenden/desaktivierenden Einfluss haben und wohin sie bevorzugt den Zweit-Substituenten hin orientieren.
Rein statistisch gesehen gibt es nach einer Erst-Substitution fünf weitere Orte für die Zweit-Substitution: zwei ortho-, zwei meta- und eine para-Stellung. Jede Position besitzt eine Wahrscheinlichkeit von 20 %, belegt zu werden. Addiert man die Positionen, erhält man zu 60% die Wahrscheinlichkeit der Belegung von ortho- und para-Stellungen und zu 40% die Belegung der meta-Position. Jede Abweichung von diesem Verteilungsmuster ist somit auf den Einfluss des Erst-Substituenten zurückzuführen. Ein Vergleich der Reaktionsgeschwindigkeiten mit der Erst-Substitution von Benzol erlaubt Rückschlüsse darüber, ob der Erst-Substituent aktivierend oder desaktivierend auf die Zweit-Substitution wirkt. Auf weitere Details bzw. konkrete Messergebnisse kann im Rahmen dieser Darlegung nicht weiter eingegangen werden.
Einfluss auf die RG Orientierung |
aktivierend |
desaktivierend |
|
ortho-/para- |
stark: -NH2 (-NHR, NHR2), -OH mäßig: -OCH3, (-OC2H5, usw) schwach: - C6H5, -CH3 (-C2H5, usw.) |
-F, -Cl, -Br, -I |
|
meta- |
|
-NO2, -N(CH3)3+, -CN, -COOH - COOR, -SO3H, -CHO, -COR |
Mit Hilfe dieser Daten lassen sich nun Reaktionen voraussagen bzw. zu einem gewünschten Endprodukt hinsteuern. Da bei der präparativen Synthese möglichst die Herstellung einer Rein-Substanz angestrebt und die Bildung von Isomeren-Gemischen vermieden werden soll wegen der anschließenden Trennungsprobleme, erlaubt die Kenntnis der Substituenteneinflüsse die Wahl der richtigen Synthesestrategie. Es macht also einen Unterschied, ob man zur Herstellung eines gewünschten Endprodukts erst den Substituenten A und dann B einführt oder umgekehrt. Soll etwa Bromnitrobenzol hergestellt werden, erhält man bei der Bromierung von Nitrobenzol das Produkt m-Bromnitrobenzol.
Soll jedoch o- und p-Bromnitrobenzol dargestellt werden, wird man Benzol zuerst bromieren und dann nitrieren, dadurch erhält man 38% o-Bromnitrobenzol und 62% p-Bromnitrobenzol. Die Synthesestrategie hängt also vom gewünschten Endprodukt ab.
Ebenso verhält es sich mit der Umwandlung von Substituenten in andere: m-Nitrobenzoesäure lässt sich durch Nitrierung von Benzoesäure erstellen, die wiederum aus der Oxidation von Toluol hergestellt werden kann. Auf diesem Wege lässt sich aber o- und p-Nitrobenzoesäure nicht erzeugen. Dagegen kann Toluol per Nitrierung in o- und p-Nitrotoluol umgewandelt werden und diese per Oxidation in o- und p-Nitrobenzoesäure. Da sich o- und p-Isomere im Siedepunkt häufig stark unterscheiden, lässt sich das Trennungsproblem per fraktionierter Destillation elegant lösen.
3. Reaktivität und Orientierung
Wenn bestimmte Substituenten den Benzolring aktivieren und eine Zweitsubstitution bevorzugt in o- und p-Stellung dirigieren, andere desaktivieren und bevorzugt in m-Stellung orientieren, dann hängt das sicher damit zusammen, was im langsamsten, geschwindigkeitsbestimmenden Schritt der elektrophilen Substitution passiert. Der langsamste, geschwindigkeitsbestimmende Schritt ist die Bildung des Carbenium-Ions, egal, welches spezifische Reagens daran beteiligt ist. Unterschiede in der Substitutionsgeschwindigkeit sollten daher auf Unterschiede in der Geschwindigkeit dieses Schrittes beruhen. Diese Unterschiede gehen bei nahe verwandten Reaktionen wiederum auf Unterschiede in der Aktivierungsenergie zurück, also auf Stabilitätsunterschiede der Übergangszustände. Wie bei anderen Reaktionen mit Carbenium-Ionen stabilisieren Faktoren, die die Energie des Carbenium-Ions durch Verteilung der positiven Ladung senken, auch das sich bildende Carbenium-Ion im Übergangszustand. Stabilere, weil energieärmere Carbenium-Ionen entstehen also leichter und sind deswegen bevorzugt. Bei der elektrophilen aromatischen Substitution ist das intermediäre Carbenium-Ion ein Resonanzhybrid aus drei Grenzstrukturen I-III, in dem die positive Ladung über den Ring verteilt ist. In o- und p-Stellung zu dem angegriffenen C-Atom ist diese positive Ladung am größten. Eine Gruppe, die bereits mit dem Benzol-Ring verbunden ist, sollte die Stabilität des Carbenium-Ions durch Verteilung oder Verstärkung der positiven Ladung entsprechend ihrem elektronenanziehenden oder -abstoßenden Charakter beeinflussen. Aus der Darstellung der Grenzstrukturen geht hervor, dass sich dieser stabilisierende bzw. destabilisierende Einfluss vor allem in den o- und p-Stellungen bemerkbar machen sollte.
4. Reaktivität
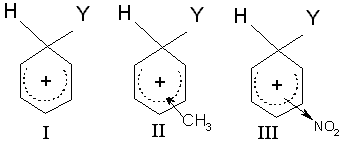
Zum Vergleich der Substitutionsgeschwindigkeiten von Benzol, Toluol (aktivierend und o-/p-dirigierend) und Nitrobenzol (desaktivierend und m-dirigierend) werden die Strukturen der Carbenium-Ionen betrachtet (Abb. 2): Die elektronenliefernde Methylgruppe (II) verringert die positive Ladung des Rings, indem sie selbst einen Teil der Ladung übernimmt. Diese Art der Ladungsverteilung stabilisiert das Carbenium-Ion und entsprechend die sich entwickelnde positive Ladung im Übergangszustand. Die Reaktion wird auf diese Weise durch den +I- (positiver induktiver) Effekt der Methylgruppe beschleunigt. Dagegen übt die NO2-Gruppe (III) einen elektronenanziehenden (-I)-Effekt aus, der die positive Ladung des Carbenium-Ions erhöht, dadurch die Stabilität vermindert und so die Reaktion verlangsamt.
Die Reaktivität bei der elektrophilen aromatischen Substitution hängt also von der Fähigkeit eines Substituenten G ab, Elektronen abzugeben oder anzuziehen.
Ein elektronenliefernder Substituent aktiviert den Benzolring, ein elektronenziehender Substituent desaktiviert den Ring.
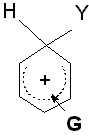
|
G ist ein Elektronendonator G stabilisiert das Carbeniumion G aktiviert |
G = -NH2, -OH, -OCH3, -NHCOCH3, -C6H5, -CH3 |
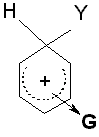
|
G ist ein Elektronenakzeptor G destabilisiert das Carbeniumion G desaktiviert |
G = -NO2, -N(CH3)3+, -CN, -COOH, - COOR, -SO3H, -CHO, -COR, -Hal |
Die Ursache der Wirkung von G als Elektronendonator oder -Akzeptor ist unterschiedlich: teils geht sie auf induktive Effekte, teils auf mesomere Effekte, teils auf beide zurück. Induktive und mesomere Effekte können sich gegenseitig verstärken oder abschwächen. Zudem spielt es eine Rolle, ob ein Substituent über freie, nichtbindende Elektronenpaare verfügt und welche Elektronegativität er besitzt.
5. Orientierung
Eine aktivierende Gruppe aktiviert alle Positionen des Benzolrings, sogar die m-Stellungen sind reaktiver als eine einzelne Position im Benzol selbst. Sie aktiviert allerdings die o- und p-Stellungen wesentlich stärker und wirkt so o-/p-dirigierend.
Eine desaktivierende Gruppe desaktiviert alle Ringpositionen, auch die m-Stellung. Sie vermindert jedoch die Reaktivität in o- und p-Stellungen stärker als in m-Stellung und dirigiert den neu eintretenden Substituenten bevorzugt in m-Stellung.
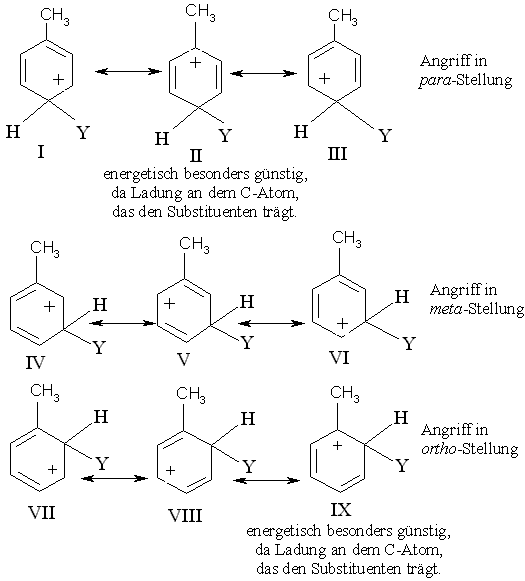
o-,m- und p-Stellung.
Mit anderen Worten: Die Orientierung in die o-, m- und p-Position kommen auf die gleiche Weise zustande, der Einfluß eines beliebigen Substituenten - aktivierend oder desaktivierend - wirkt sich jedoch in den o- und p-Stellungen am stärksten aus.
Vergleicht man die Grenzstrukturen beim Angriff eines Elektrophils auf das Toluol (aktivierende Methylgruppe) in den drei Stellungen miteinander (Abb. 3), so wird deutlich, dass die positive Ladung in Struktur II deswegen am stärksten erniedrigt wird, weil sie direkt an dem C-Atom sitzt, das den elektronenliefernden Substituenten Methylgruppe trägt. Die gleiche Ladungsverteilung findet sich in Struktur IX (Angriff in o-Stellung). Die CH3-Gruppe gibt zwar an alle Ringpositionen Elektronen ab, doch am meisten an das ihr benachbarte C-Atom. Deswegen sind die Strukturen II und IX besonders stabil. Das dem Angriff auf die o- bzw. p-Stellung entstammende Carbenium-Ion ist wegen der Beteiligung der Strukturen II bzw. IX am Resonanzhybrid stabiler als das Carbenium-Ion, das sich bei einem Angriff auf die m-Stellung bildet. Deshalb erfolgt die o-/p-Substitution rascher als die m-Substitution.
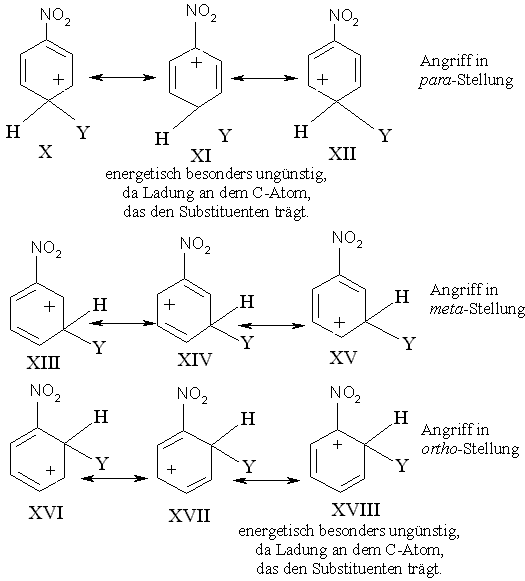
Vergleicht man nun damit die Carbenium-Ionen-Strukturen, die beim Angriff eines Elektrophils auf das Nitrobenzol (desaktivierende Gruppe) formuliert werden können (Abb. 4), so wird deutlich, dass bei den Grenzstrukturen XI und XVIII die positive Ladung gerade an dem C-Atom sitzt, das mit der elektronenziehenden NO2-Gruppe direkt verbunden ist. Die NO2-Gruppe zieht zwar von allen Ringpositionen Elektronen ab, aber am stärksten von ihrem Nachbarkohlenstoffatom. Deswegen zeigt dieses C-Atom, das wegen des elektronenziehenden Effekts schon positiv polarisiert ist, nur wenig Neigung, die positive Ladung des Carbenium-Ions zu übernehmen. Die Strukturen XI und XVIII sind daher besonders instabil und tragen wenig dazu bei, das aus dem p- oder o-Angriff resultierende Carbenium-Ion zu stablisieren. Das Ion des p-Angriffs ist letztlich ein Resonanzhybrid aus nur zwei Strukturen X und XII bzw. XVI und XVII bei einem Angriff in o-Stellung, die positive Ladung ist hauptsächlich auf nur zwei C-Atome beschränkt. Das Ion des p- bzw. o-Angriffs ist deswegen weniger stabil als das bei einem Angriff auf die m-Stellung entstehende Carbenium-Ion, das ein Resonanzhybrid dreier Strukturen ist, und bei dem die positive Ladung von drei C-Atomen übernommen wird, die nicht direkt mit der elektronenziehenden NO2-Gruppe verbunden sind. Aus diesem Grund verläuft die o-/p-Substitution langsamer als die Substitution in m-Stellung.
Bei Nitrobenzol vollzieht sich also die o-/p-Substitution langsamer als die m-Substitution, denn der elektronenziehende Effekt der NO2-Gruppe wirkt sich bei einem o-/p-Angriff stärker desaktivierend aus.
Damit wird klar, dass sowohl die o-/p-Orientierung bei den aktivierenden Substituenten wie auch die m-Orientierung bei den desaktivierenden Substituenten logisch aus der Struktur des intermediären Carbenium-Ions folgt: Die Ladung ist in den Positionen ortho und para zum angegriffenen C-Atom am stärksten und deshalb erzielt sowohl ein aktivierender als auch ein desaktivierender Substituent in diesen Stellungen den größten Effekt.
Das ungewöhnliche Verhalten der Halogene, die o-/p-dirigierend und zugleich desaktivierend sind, resultiert aus zwei einander entgegenwirkender Faktoren und wird später erläutert.
Substituenten beeinflussen also durch ihre Fähigkeit, Elektronen abzugeben oder anzuziehen sowohl die Reaktivität als auch die Orientierung einer elektrophilen aromatischen Zweit-Substitution. Die bisher dargestellten Effekte sind induktive Effekte: sie gehen zurück auf die Elektronegativität des betreffenden Substituenten.
Manche Gruppen jedoch wie -NH2 oder -OH bzw. deren Derivate wirken aktivierend, obwohl die elektronegativere Atome als das C-Atom enthalten und einen elektronenziehenden, also -I-Effekt ausüben und somit desaktivierend wirken sollten. Diese Gruppen müssen also noch eine zweite Möglichkeit besitzen, Elektronen an das Ringsystem abzugeben, da ihre Wirkung ja aktivierend ist. Diese zweite Möglichkeit beruht auf der Überlagerung freier nichtbindender Elektronenpaare des Substituenten mit den π-Elektronen des aromatischen Systems und wird als Resonanz- oder Mesomerie-Effekt bezeichnet. Das Stickstoff-Atom, obwohl elektronegativer als das C-Atom, wirkt (im Sinne der Lewis-Säure-Basen-Theorie) basisch: es neigt dazu, sein freies, nicht bindendes Elektronenpaar einem anderen Atom zur Verfügung zu stellen und eine positive Ladung zu übernehmen. Genauso wie Ammoniak NH3 unter Bildung des Ammonium-Ions NH4+ ein Proton anlagert, addieren dem Ammoniak verwandte Verbindungen Protonen und gehen in substituierte Ammonium-Ionen über.
Das Reaktionsprinzip |NR3 + H —> NHR3+ mit R = H oder C-Verbindungen formuliert diese Reaktion.
Die Hydroxyl-Gruppe besitzt eine ähnliche, wenn auch schwächere Basizität (im Sinne von Lewis). Hier verläuft die Reaktion nach dem Prinzip H2 O| + H+ –> H3 O+, wobei auch hier H durch C-Atome ersetzt werden kann.
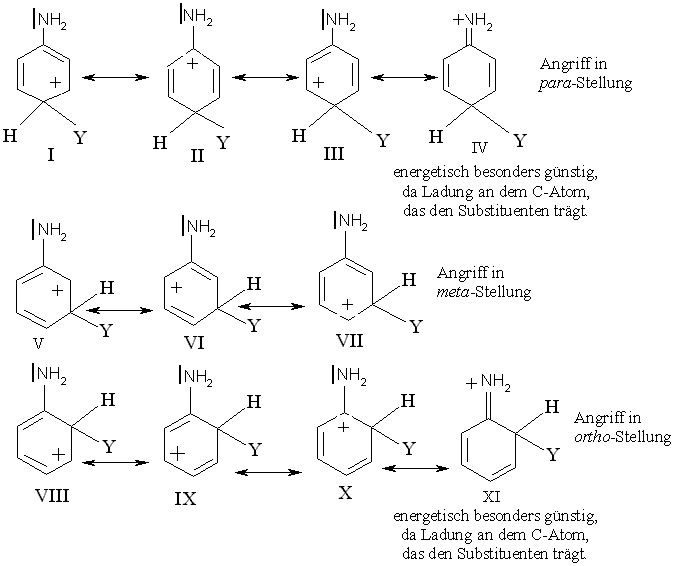
Der aktivierende Einfluss der -NH2 und -OH-Gruppe lässt sich die Annahme erklären, dass Stickstoff und Sauerstoff mehr als ein Elektronenpaar mit dem aromatischen Ring teilen und dadurch eine positive Ladung aufnehmen können.
Das Carbenium-Ion, das durch einen Angriff in p-Stellung zur NH2-Gruppe des Anilins entsteht, stellt nicht nur ein Resonanzhybrid der Strukturen I-III dar, in denen die positive Ladung auf den C-Atomen des aromatischen Rings ruht, sondern bezieht auch die Struktur IV ein, in der das N-Atom die positive Ladung trägt. Die Struktur IV ist besonders stabil, da in ihr jedes Atom (außer H) ein vollständiges Elektronenoktett besitzt. Das Carbenium-Ion ist wesentlich stabiler als das bei einem Angriff auf Benzol entstehende Ion oder als die Ionen, die sich bei einem Angriff auf die m-Position zur NH2-Gruppe des Anilins (V- VII) bilden können. In keinem dieser Fälle ist eine Struktur wie IV möglich. Der Angriff in der o-Position mit den Strukturen VIII - XI gleicht annähernd dem Angriff auf die p-Position. So erfolgt die Substitution des Anilins schneller als die des Benzols und bevorzugt in o- und p-Stellung zur NH2-Gruppe.
In der gleichen Weise kann man die aktivierende und o-/p-dirigierende Wirkung der OH-Gruppe durch Beteiligung von Strukturen wie XII und XIII deuten, in denen wieder jedes Atom ein vollständiges Elektronenoktett besitzt. Für Derivate der NH2- und OH-Gruppe gelten dieselben Überlegungen, sind es doch die freien nichtbindenden Elektronenpaare des N- und O-Atoms, die diesen Resonanzeffekt zustandekommen lassen.
6. Halogene
Die Halogene sind eine ungewöhnliche Substituentengruppe: sie wirken desaktivierend und dennoch o-/p-dirigierend. Für
elektronenanziehende Substituenten (-I-Effekt) ist die erste Wirkung verständlich, die zweite Wirkung der o-/p-Direktion ist jedoch ein Kennzeichen elektronenliefernder Substituenten. Können Halogene also beides: Elektronen anziehen und gleichzeitig abgeben? Die Antwort lautet ja, nur die Ursachen sind verschieden. Ein Halogenatom kann über den induktiven Effekt Elektronen anziehen und über den Mesomerieeffekt Elektronen abgeben. Vermutlich ist bei der NH2- und OH-Gruppe dies auch der Fall, jedoch überwiegt der Resonanzeffekt den induktiven Einfluss. Bei den Halogenen wirken sich die beiden gegenläufigen Effekte ungefähr in gleichem Maße aus.
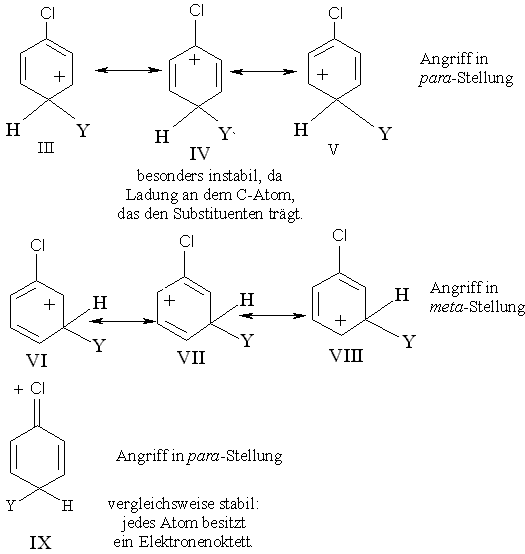
Bei einem elektrophilen Angriff auf z.B. Chlorbenzol entsteht ein Carbenium-Ion, wobei Cl als Erstsubstituent durch seinen elektronenziehenden Effekt (-I) die positive Ladung des Carbenium-Ions verstärkt, wodurch das Ion instabiler und die Reaktion insgesamt langsamer wird.
Um die Orientierungswirkung verstehen zu können, werden Grenzstrukturen verglichen, die aus einem Angriff in m- und p-Stellung resultieren. Jedes dieser Ionen ist ein Resonanzhybrid dreier Strukturen III - V für die p- und VI-VIII für die m-Substitution. Bei der Struktur IV ist die positive Ladung gerade an dem C-Atom lokalisiert, an dem sich auch das Cl-Atom befindet. Aufgrund seines induktiven Effektes zieht Chlor von dem C-Atom, mit dem es verknüpft ist, Elektronen ab, weshalb die Struktur IV besonders instabil ist. IV sollte daher nur geringfügig am Resonanzhybrid beteiligt sein, das daher weniger stabil sein sollte als das Resonanzhybrid aus dem m-Angriff. Der induktive Effekt alleine würde also nicht nur die Desaktivierung, sondern auch die m-Orientierung bewirken.
Halogenatome können jedoch aufgrund ihrer freien, nichtbindenden Elektronenpaare auch einen Mesomerie-Effekt ausüben. Durch die Teilung eines Elektronenpaars mit einem anderen Atom übernehmen sie eine positive Ladung. Das bedeutet, dass das Carbenium-Ion, das bei einem Angriff in p-Stellung entsteht, ein Resonanzhybrid nicht nur der Strukturen III - V ist, sondern auch der Struktur IX, in der das Cl-Atom eine positive Ladung trägt und durch eine Doppelbindung an den Ring gebunden ist. Diese Struktur sollte vergleichsweise stabil sein, weil alle Atome aus H ein Elektronenoktett besitzen. Die Struktur IX entspricht genau denselben Strukturen, die man für die aktivierende und o-/p-dirigierende Wirkung der NH2- und OH-Gruppe verantwortlich macht. Bei einem Angriff in m-Stellung entsteht kein derartiges Ion. Das Ion des p-Angriffs ist um so stabiler im Vergleich zu einem Angriff in m-Stellung, je größer die Beteiligung der Struktur IX am Resonanzhybrid ist. Obwohl man nichts über die relative Bedeutung der zwei Effekte - geringe Stabilität der Struktur IV und die Stabilisierung durch IX - aussagen kann, deuten die experimentellen Ergebnisse darauf hin, dass die Resonanzbeteiligung durch die Struktur IX ausschlaggebend ist.
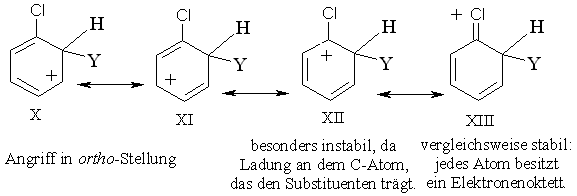
In ähnlicher Form wird das Ion (X - XIII), das bei einem Angriff in o-Stellung entsteht, dadurch stabilisiert, dass das Cl-Atom eine positive Ladung übernimmt.
Das Halogenatom zieht durch seinen -I-Effekt Elektronen aus dem Ring, dadurch wird das intermediäre Carbenium-Ion instabiler. Dieser Einfluss tritt bei einem Angriff auf alle Ringpositionen in Erscheinung, wirkt sich jedoch in o- und p-Stellung zum Halogen am stärksten aus. Das Halogen liefert aber durch seine freien Elektronenpaare einen Beitrag zum Resonanzeffekt, der Elektronen an den Ring liefert. Dadurch wird das intermediäre Carbenium-Ion stabilisiert. Dieser Effekt wird jedoch nur bei einem Angriff in o- und p-Position wirksam. Der induktive Effekt überwiegt den Resonanzeffekt und verursacht insgesamt einen Elektronenzug - er wirkt somit desaktivierend in allen Ringpositionen. Der Resonanzeffekt ist bei einem Angriff auf die o- und p-Stellungen dem induktiven Effekt entgegen gerichtet und damit werden die o- und p-Stellungen weniger desaktiviert als die m-Stellung. So bestimmt der stärkere induktive Effekt die Reaktivität und der schwächere Resonanzeffekt die Orientierung.
Literatur: Morrison/Boyd: Lehrbuch der Organischen Chemie, VCH, Weinheim 1974; Abschnitt 11.2 bis 11.21, leicht verändert und gekürzt.
update am: 02.02.21 zurück zur Hauptseite